






Machine Learning enhanced with Quantum Chemistry
The equations of quantum mechanics provide a roadmap to predicting the properties of chemicals starting from basic scientific theories.
However, these equations quickly become too expensive in terms of computer time and power when used to predict behaviour in large systems. Machine learning offers a promising approach to accelerating such large-scale simulations.
Researchers have shown that machine learning models can mimic the basic structure of the fundamental laws of nature. These laws can be very difficult to simulate directly. The machine learning approach enables predictions that are easy to compute and are accurate in a wide range of chemical systems.
The Impact
The improved machine learning model can quickly and accurately predict a wide range of properties of molecules. These approaches score very well on important benchmarks in computational chemistry and show how deep learning methods can continue to improve by incorporating more data from experiments.
The model can also succeed at challenging tasks such as predicting excited state dynamics—how systems behave with elevated energy levels. This tool is a breakthrough capability for quantum chemistry. It will allow researchers to better understand the reactivity and excited states of new molecules.
Summary
The use of machine learning to predict chemical properties offers the promise of great technological advances, with applications from cleaner energy to faster pharmaceutical drug design.
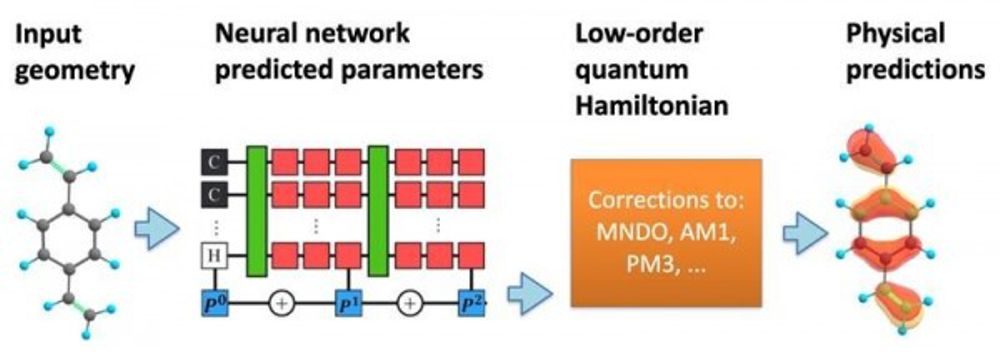
This is a highly active area of research, but most existing approaches use simple and heuristic approaches to the design of the machine learning models. In this new work, researchers from Los Alamos National Laboratory have proposed incorporating more of the mathematics of quantum mechanics into the structure of the machine learning predictions.
Using the specific positions of atoms within a molecule, the machine learning model predicts an effective Hamiltonian matrix, which describes the various possible electronic states along with their associated energies.
Compared to traditional quantum chemistry simulations, the machine learning-based approach makes predictions at a much-reduced computational cost. It enables quantitatively precise predictions regarding material properties, allows interpretable insight into the nature of chemical bonding between atoms, and can be used to predict other complex phenomena, such as how the system will respond to perturbations, such as light-matter interactions.
The method also provides greatly improved accuracy relative to traditional machine learning models, and demonstrates success in transferability, i.e., the ability of the model to make predictions that go well beyond the data that formed the basis of its training.
Funding
This research was supported by funds from the Los Alamos National Laboratory’s Laboratory Directed Research and Development program and by the Department of Energy Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division.
This work was performed in part at the Center for Nonlinear Studies and the Center for Integrated Nanotechnology, a DOE Office of Science user facility.




















